

Cryopreservation is the process by which cells, tissues or organs are cooled and stored at very low or freezing temperatures to save them for future use. The cryopreservation of equine embryos has always been a desirable addition to modern breeding technologies because it provides breeders with the option to bank embryos of known or potential genetic merit. Hence, young mares who have not yet proven themselves can have embryos cryopreserved for future transfer when the donor mare shows value, or older stock of merit can produce offspring from banked embryos beyond their reproductive lifespan. The technique of cryopreservation also acts as a management tool by allowing embryos to be kept and transferred when a suitable recipient is available or to transfer them at a time of the year that will ensure foalings occur at the desired time. In addition, cryopreserved embryos can be moved internationally, increasing both the availability of bloodlines and worldwide trade between breeders.
The challenges of cryopreserving equine embryos
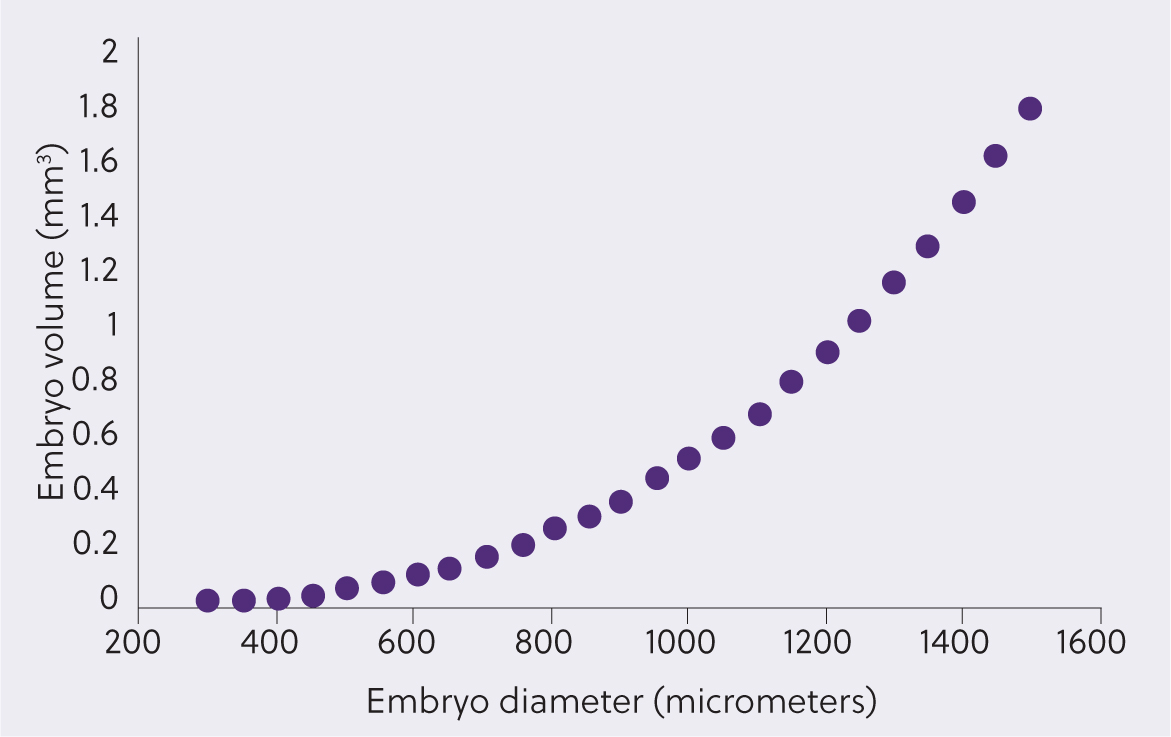
Mammalian embryos cryopreserve well when they are relatively small, contain a negligible blastocoele cavity and their structure allows for the passage of cryoprotectants into the cells. Equine embryos have many unique features which challenge these requirements (Figure 1). These include the long oviducal transport time of the equine embryo of 6–6.5 days (Battut et al, 1997), coupled with its rapid blastulation and, hence, increase in volume once it arrives in the uterus as it develops from a morula to early blastocyst (Figure 1), making the recovery of small embryos difficult. The volume of the blastocoele, and therefore the amount of fluid within an embryo, also increases exponentially in relation to its diameter (Figure 2), meaning both the risk of ice formation and the distance of diffusion of cryopreservation media into the embryo increases as it enlarges. To further add to the challenge of cryopreserving equine embryos, they also develop a tough acellular glycoprotein coating, the so-called blastocyst capsule, underneath the zona pellucida (Betteridge, 1989; Oriol et al, 1993; Figure 1). This capsule is believed to limit the passage of cryoprotectants into the embryo (Pfaff et al, 1993; Legrand et al, 2000; Gillard Kingma et al, 2011). Finally, from around day 7 the blastocoele cavity changes into a two-layered yolk sac (bilaminar omphalopleure) with formation of the endoderm (Enders et al, 1993). This additional endoderm layer within the embryo may also hinder the passage of cryoprotectants and render it more vulnerable to damage during cryopreservation.

Hence, recovery of small embryos that cryopreserve well has historically meant flushing mares on day 6–6.5. However, without an accurate timing of ovulation, embryo recovery rates can be lower than those achieved from flushing on day 7 (Boyle et al, 1989).
The rising use of intracytoplasmic sperm injection in Europe and beyond has dramatically increased the number of equine embryos that have been cryopreserved (Stout, 2020). However, it must be remembered that these in-vitro produced embryos with their smaller diameter, smaller blastocoele cavity and poorly developed capsule are considerably easier to freeze than in vivo produced ones. This review only deals with the vitrification of in vivo produced embryos, although cryopreservation techniques are similar for the two types of embryos.
Overview of cryopreservation methods
The cryopreservation of living cells and tissues presents some problems that need to be overcome to successfully freeze them. These are chiefly related to the way water, a ubiquitous component of cells, reacts as the temperature drops. Water within cells acts as a solvent to dissolve and ‘carry’ other molecules and as the temperature approaches freezing, water molecules gather together to form ice crystals. This causes two potential problems for the cell. First, the ice pushes other molecules into a potentially harmful concentrated solution and, second, as a result of the expansion of water when it freezes, physical damage can occur to the cells. To overcome these problems cryoprotectants are used to help protect cells during cryopreservation.
Cryoprotectants are described as either penetrating or non-penetrating, depending on their ability to pass into the cell or remain in the extracellular environment. Their helpful effects are conveyed primarily through interactions with or the manipulation of intracellular and extracellular water. Non-penetrating cryoprotectants are molecules that are too large to diffuse into cells. They exert their beneficial effects by increasing the osmolarity of extracellular fluid, thus helping to increase cellular dehydration and therefore reduce ice crystal formation within the cells. Examples of non-penetrating cryoprotectants are sugars, such as sucrose or trehalose, and macromolecules, such as ficoll and polyethylene glycol. These molecules are also often used in media when thawing or warming cryopreserved cells to prevent a sudden influx of water and potential osmotic shock (Swain and Smith, 2010).
In contrast, penetrating cryoprotectants are small, non-ionic compounds with a high solubility in water at low temperatures; examples used in the cryopreservation of equine embryos include ethylene glycol, dimethyl sulfoxide and glycerol. These substances can diffuse across cell membranes, although depending on the cryoprotectant this may take some time. The theory is that they replace most of the intracellular water to prevent excessive cell dehydration while preventing ice formation. Their success is often related to the speed at which they can cross the cell membrane. Since penetrating cryoprotectants solidify at lower temperatures than water, they reduce ice formation at any given temperature, thus helping to prevent the physical damage that ice can cause to organelles and cell membranes (Swain and Smith, 2010). However, these compounds are toxic at high concentrations or if exposure is prolonged before cryopreservation. Combinations of penetrating cryoprotectants are often used, as the toxicity of lower concentrations of two different cryoprotectants is considered less than a higher concentration of one (Fahy, 2014; Fahy and Wowk, 2015). Combinations used to cryopreserve equine embryos have included ethylene glycol and dimethyl sulfoxide or ethylene glycol and glycerol.
Although this review focuses on the vitrification of embryos, it is useful to know that there are two different methods used for the cryopreservation of equine embryos – these are referred to as slow freezing and vitrification.
Slow freezing uses low levels of cryoprotectants and slow rates of freezing (0.3–0.5 °C/min). The slow dehydration that occurs during freezing aims to minimise ice formation. Typically, embryos are loaded into 0.25 ml straws with cryoprotectant solutions and placed in a programmable freezing machine. The temperature is dropped slowly and at -5 °C, an ice nucleation induction step called manual seeding is undertaken before further slow cooling and eventual plunging into liquid nitrogen. The whole procedure takes many hours and needs specialist equipment, but is relatively easy to do.
Vitrification is not freezing, but the transformation of a sub-stance into a glass-like, non-crystalline, amorphous solid. It relies on exposure of the embryo to high levels of cryoprotectants for relatively short times and a very fast cooling rate (approximately 20 000 °C/min) which in theory should prevent the formation of ice crystals as the kinetics of ice crystal formation are ‘outrun’. Embryos are plunged into liquid nitrogen after being loaded onto minimal vitrification devices in <1 µl of cryoprotectant. Warming of vitrified embryos (note that this is not referred to as thawing) is also undertaken very quickly to prevent devitrification – a process where ice crystal formation occurs during warming. No specialist equipment is required, and the vitrification process takes minutes to undertake (Vanderzwalmen et al, 2016). However, despite being a relatively easy method for cryopreservation, some of its effectiveness can be influenced by many factors which are summarised in Table 1.
Table 1. Factors that can profoundly influence the effectiveness of vitrification.
| Type and concentration of cryoprotectant (almost all cryoprotectants are toxic) |
| Media used as a base medium (holding media) |
| Temperature of the vitrification solution at exposure |
| Length of time cells or tissue are exposed to the final cryoprotectant before being plunged into liquid nitrogen |
| Variability in the volume of cryoprotectant solution surrounding the cells or tissue |
| Device used for vitrification (influences size of the liquid nitrogen vapour coat and cooling rate) |
| Technical proficiency of the embryologist (‘the learning curve’) |
| Quality and developmental stage of the cell or tissue (very pertinent to equine embryos) |
| Direct contact of the liquid nitrogen and the vitrification solution containing the biological material can be a source of contamination (sterile liquid nitrogen should be used for cooling or storage or closed vitrification devices) |
Historical aspects of equine embryo vitrification
The path to the present situation where in vivo equine embryos can be vitrified with high success rates has been a long one. Hochi et al (1994) were the first authors to report successful pregnancies from vitrified equine embryos, although pregnancy rates were far from ideal (2 out of 5; 40%). However, the same research group subsequently reported on the developmental competence in vitro of vitrified embryos on the basis of their size and found that only 25% of embryos >300 µm vs 81% of those ≤300 µm developed in vitro after vitrification and warming, illustrating that as with slow freezing, larger equine embryos were more prone to damage following vitrification compared to smaler embryos (Hochi et al, 1995). Some 10 years later, Eldridge-Panuska et al (2005) demonstrated high success rates when vitrifying embryos using a commercially practical protocol. Embryos were exposed to a three-step protocol using solutions containing increasing concentrations of ethylene glycol and glycerol before being loaded into 0.25 mL straws which were suspended over liquid nitrogen for 1 minute before being plunged into it. Warming was also very simple using a two-step dilution method.
In the initial study, transfer of six morulae or early blastocysts ≤300 µm resulted in four pregnancies (four out of six; 67%), although no pregnancies occurred when embryos >300 µm were transferred. The warming protocol was subsequently further simplified allowing mixing of the dilution solution within the straw and direct transfer, with pregnancy rates using this technique still commercially acceptable at 62% (16 out of 26). This research was translated into a commercial vitrification kit. Despite excitement at its availability and the promise of being able to vitrify embryos with a reasonable success rate, it is worth pointing out that the embryos needed to be carefully selected to optimise post warming survival rates. Hence, ideally, embryos needed to be ≤ 250 µm in diameter and, if already blastulating, the zona pellucida needed to be no more than half its normal thickness. Despite the challenges of obtaining such small embryos when flushing, embryos vitrified using this technique have produced acceptable pregnancy rates in the field (59%; 38 out of 64; Araujo et al, 2010). Nevertheless, the desire to be able to cryopreserve embryos >300 µm persisted.
Vitrification techniques for equine embryos >300 µm
Although reports of the transfer of larger vitrified embryos existed (Caracciolo di Brienza et al, 2004), initially none of the recipient mares established a pregnancy. Some minor improvement was described by Campos-Chillon et al (2006) when an embryo survival rate of 35% (6 out of 17) was reported, although embryos >400 µm failed to form pregnancies.
It was a few more years before a significant leap forward was made in vitrifying larger embryos. Although not working primarily on embryo vitrification, Choi et al (2010) punctured the embryonic capsule and collapsed the trophoblast while obtaining trophoblast biopsies for pre-implantation genetic diagnosis. Subsequent transfer of these embryos resulted in pregnancies in recipient mares, showing that puncture and trophoblast collapse was compatible with embryo survival. This led the same authors to subsequently vitrify expanded blastocysts (407–565 µm) similarly treated, resulting in a pregnancy rate of 71% (five out of seven; Choi et al, 2011). With varying vitrification protocols, but all relying on the puncture and aspiration of the blastocoele, several other groups achieved similar pregnancy rates (Diaz et al, 2016; Sanchez et al, 2017; Wilsher et al, 2019), with embryos that formed pregnancies ranging in size from 353–1200 µm.
Although it is tempting to attribute the success of these methodologies to the aspiration and puncture of the embryos, it is likely that vitrification per se and the use of minimal volume devices (Figure 3) on which embryos are vitrified play important roles in the improved pregnancy rates for larger embryos. Evidence for this comes from a recent paper which demonstrated that vitrification compared to slow freezing was less damaging to embryos, even though protocols for both experimental cryopreservation methods used collapse and blastocoel aspiration as a pre-treatment (Umair et al, 2023). Furthermore, vitrification of collapsed embryos larger than 300 µm on either a hemi-straw, a minimum vitrification volume device, or within a Stripper-tip holding a larger volume demonstrated that a minimal vitrification volume was essential for embryo viability post warming with 70% (7 out of 10) vs 0% (none out of five) pregnancy rates respectively (Sanchez et al, 2017). Such minimal volume devices are used routinely and to good effect to vitrify human embryos (Scholz, 2021). Their advantages come from the fact that they allow for increases in cooling and warming rates with lower cryoprotectant concentrations, which help to reduce the detrimental effects of toxicity and osmotic shock to the cells (Ghetler et al, 2005; Katkov et al, 2007).

The initial reports of successful vitrification of large equine embryos (>300 µm) all used collapse of the blastocoele cavity by its puncture and aspiration, with this being achieved using a micromanipulator mounted on an inverted microscope (Figure 4; Choi et al, 2011; Diaz et al, 2016; Sanchez et al, 2017; Wilsher et al, 2019). It was suggested in these previous reports that 85–95% of the blastocoele fluid should be aspirated to ensure the embryo was as collapsed as possible before vitrification. However, a study undertaken to determine if this was really the case showed that puncture without aspiration gave equivalent pregnancy rates to embryos that were punctured and aspirated (80% (8 out of 10) vs 75% (9 out of 12); P=0.82, respectively) provided the embryos were ≤560 µm in diameter. For embryos bigger than 560 µm in diameter, there was a clear advantage to aspirating them (aspirated 72% ((13 out of 18) versus non-aspirated 10% (1 out of 10); P=0.006); (Wilsher et al, 2019). This work provided the ground-work for devising more user-friendly protocols.

Making vitrification easier to accomplish
Although Wilsher et al (2019) successfully vitrified embryos without aspiration, the embryos still required puncture. To accomplish this, a microscope mounted micro-manipulator was required. This is an expensive piece of equipment requiring some user skill to master, so a simpler methodology is required. To this end, manual puncture methods have been reported. For example, Guignot et al (2016) manually punctured 28 expanded blastocysts (166–777 µm in diameter) with a glass pipette before collapsing them further by placing them in medium containing increasing sucrose concentrations before vitrification on open pulled straws. Then, 25 embryos were warmed and cultured in vitro with the authors reporting a survival rate of 96% (24 out of 25) after 24 hours. However, no embryos were transferred to test their viability in vivo. Manual collapse was also reported by Ferris et al (2016) using a 25 G hypodermic needle to puncture 15 expanded horse blastocysts (mean diameter 663 µm) before vitrification; this resulted in a reported 46% (7 out of 15) pregnancy rate after warming and transfer. However, the tip of a 25 G needle is exceptionally large compared to a blastocyst and considerable skill would be needed to avoid irreversibly damaging the embryo. Furthermore, this author has failed to establish pregnancies in non-vitrified embryos when puncture was undertaken with anything wider than 50 microns. Even puncture with a fine facial acupuncture needle (100 µm shaft) is not compatible with embryo survival (Wilsher et al, 2020). However, Wilsher et al (2021) reported manual puncture with a microprobe needle before vitrification which resulted in pregnancy rates of 82% (14 out of 17), provided embryos were ≤560 µm in diameter. Larger embryos did not survive. However, it should be noted that these manual techniques require a steady hand and some practice to achieve consistent results.
As a result, modification of protocols to allow vitrification of embryos >300 µm without puncture were required. A recent report suggests this is now possible, with non-punctured vitrified embryos between 300 and 480 µm in diameter resulting in a good pregnancy rate (75%; 12 out of 16) following warming and transfer to recipients (Kovacsy et al, 2023). Although, to date, this method has not been tested in a commercial setting. It is hoped future research will potentially make vitrification possible for even larger non-punctured embryos.
Typical vitrification and warming protocols
Although protocols do vary, typically, after puncture of the blastocoele cavity (with or without aspiration of the fluid), embryos are first exposed in one or two steps to equilibration medium (for example, in one step protocols: holding medium + 7.5% ethylene glycol + 7.5% dimethyl sulfoxide) for approximately 6 minutes before being moved to a vitrification solution (for example, holding medium + 15% ethylene glycol + 15% dimethyl sulfoxide) for around 60 seconds. During these 60 seconds, they are loaded onto a minimal volume device, such as a Cryolock (Figure 3) capped and plunged into liquid nitrogen. The vitrified embryos are warmed by uncapping the Cryolock and immediately plunging the tip of the device into 1 ml of warming solution 1 (for example, holding medium + 1 M sucrose) pre-warmed to 38 °C. After 1 minute the embryo is moved to a 50 µl droplet of warming solution 2 (for example, holding medium + 0.5 M sucrose) for 4 minutes at room temperature, before a final move to holding medium before transfer to a recipient mare that has ovulated 5 or 6 days previously.
Although vitrification and warming media can be made in-house, many people may lack the skills or clean areas (laminar flow hoods) in which to prepare it. Commercial media is produced for use with equine embryos, but human vitrification kits such as those from Kitazato (Kitazato Corporation, Japan) and Vit Kit (FujiFilm, Irvine Scientific Inc, USA) perform equally as well with equine embryos (Wilsher et al, 2020; Kovacsy et al, 2023).
The recently published protocol for non-punctured embryos gave good pregnancy rates (75%; 12 out of 16) when embryos between 300–500 µm were exposed to an extended time in equilibriation medium of 15 minutes and ≤90 seconds in vitrification solution (Kovacsy et al, 2023). Warming was undertaken as described above but the first warming solution was warmed to 42 °C before use. This study used the media from Kitazato.
Conclusions
Good pregnancy rates can now be achieved following the vitrification of large equine embryos, but it must be remembered that smaller embryos have higher pregnancy rates following vitrification than larger ones. Recovery of embryos from donor mares early on day 7 post-ovulation means the vast majority of embryos will be <500 µm, opening up more options for non-puncture vitrification protocols. Optimising the protocols and making them easier to accomplish without the need for costly equipment will help cryopreservation to be used increasingly as a valuable tool in modern reproductive technologies, and will increase the export market for equine embryos.
KEY POINTS
- Cryopreservation of equine embryos was historically challenging.
- Vitrification of equine embryos now results in high pregnancy rates post warming and transfer.
- Equine embryos ≤300 µm require no special treatment before vitrification.
- Equine embryos >300 µm require collapse of the blastocoele cavity before vitrification.
- Newer protocols are being developed to allow vitrification of embryos >300 µm without such pre-treatments.